







The 510(k) review process is a critical step for medical device manufacturers seeking FDA clearance. Addressing the identified deficiencies thoroughly and promptly is essential for obtaining FDA clearance for a medical device. Ensuring compliance with all regulatory requirements and providing comprehensive responses to any FDA requests will facilitate a smoother review process. In this blog, we’ll explore the types of substantive comments commonly encountered during 510(k) reviews, provide tips to avoid common pitfalls, and offer strategies for responding to comments effectively.

What is US FDA 510(k) AI Requests?
FDA issues a request for additional information (AI request) when the 510(k) submission lacks the information necessary for the Agency to complete its review and to determine whether the device is SE or NSE. AI requests are issued by email with an attached document identifying deficiencies. These requests inform the submitter that the 510(k) is being placed on hold pending receipt of a complete response to all the identified deficiencies. The hold starts on the issue date of the AI request.
An AI request is an interim action that stops the review clock and marks the end of an FDA review cycle. The review clock will resume upon the receipt of a complete response to the AI request. FDA generally issues an AI request when FDA believes the additional information needed from the submitter is not suitable for interactive review and/or cannot be provided within a reasonable timeframe (i.e., such that the review would be unduly delayed if the submission were not placed on hold).
Understanding Substantive Comments
Substantive comments from the US FDA during 510(k) reviews can cover various aspects of the submission, including technical adequacy, substantial equivalence, risk assessment, labeling, clinical data, biocompatibility, manufacturing processes, software validation, and post-market surveillance.
In the context of the FDA’s 510(k) premarket notification process, review comments can be categorized into major and minor comments based on their impact on the submission and the necessary actions to address them. Here’s a breakdown of these categories:
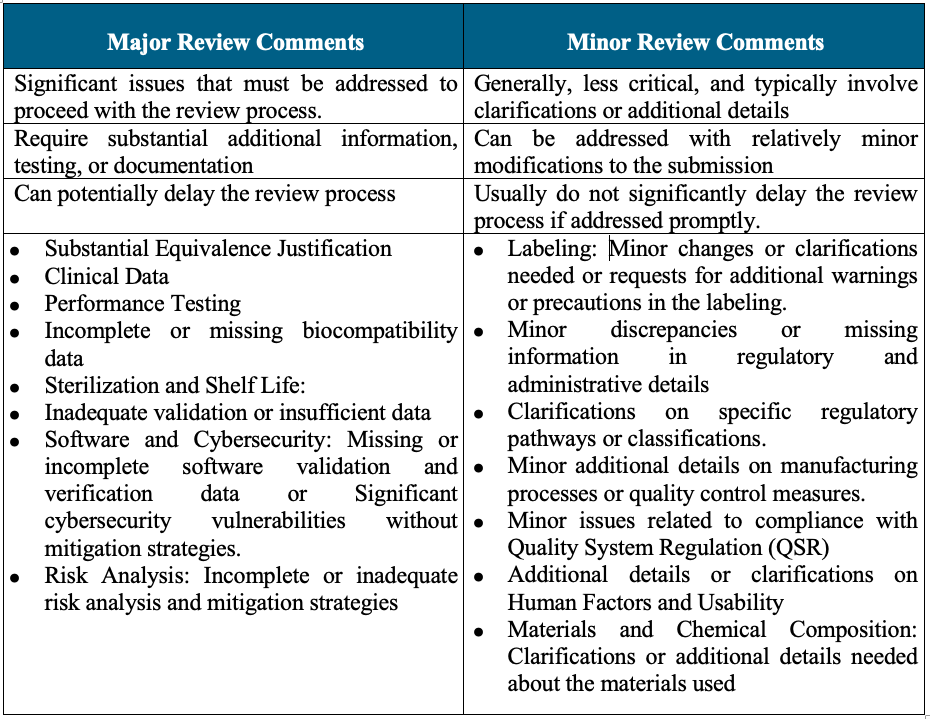
Major comments require significant attention and additional information to resolve, while minor comments generally involve clarifications or small modifications. Addressing both types of comments thoroughly and promptly is essential to ensure a smooth and timely review process for the 510(k) submission.
Response to an AI Request
The submitter should provide a complete response to an AI request from FDA. The response should address all of the deficiencies identified by FDA in its AI request to be considered a complete response. The submitter’s submission of a response to an AI request is an action that, upon receipt by FDA, resumes the review clock (i.e., the 90-day review clock resumes upon receipt of the additional information).

If FDA determines that the submitter has not addressed one or more of the deficiencies identified in the AI request, the review cycle will be terminated until FDA receives a response addressing the remaining deficiencies. In such a case, FDA informs the submitter by email that the response is incomplete and the 510(k) will be placed back on hold as of the date of the original AI request; therefore, the review clock has not resumed. The submitter will have 180 calendar days from the date of the original AI request in which to submit a complete response, or the entire 510(k) will be considered to be withdrawn.
Encountering substantive comments during 510(k) reviews is common, but with careful preparation and effective response strategies, manufacturers can navigate the process successfully. By understanding the types of comments, avoiding common pitfalls, and responding to comments in an organized and thorough manner, manufacturers can enhance the likelihood of obtaining FDA clearance for their medical devices.
For expert guidance and assistance with 510(k) submissions, contact 8C Healthcare today at contact@8chealthcare.com
Contact 8C Healthcare for expert guidance on 510(k) submissions and regulatory compliance. Let us help you navigate the path to FDA clearance.



